FDA under the influence – 6 things you should know about pharma and the FDA

“The U.S. Food and Drug Administration is supposed to protect Americans from harmful drugs. But in reality, FDA-approval does not guarantee safety. Critics say Big Pharma funds FDA reviews of new drugs, creating a conflict of interest. The agency is too focused on approving drugs to appease Big Pharma and it lacks the proper authority and funding to protect the public.”
Misplaced Trust: Why FDA Approval Doesn't Guarantee Drug Safety
In “Doctors under the influence,” we showed some of the ways in which the pharmaceutical industry manipulates doctors to influence their prescribing practices, starting with when they are students in medical school. Drug companies, like all corporations, are responsible to their shareholders. In the pharmaceutical industry, huge profits come before patients' welfare so they have no compunction about marketing drugs that have little value for most patients and drugs that they know are harmful. They view the billions of dollars in criminal and civil fines levied by the Department of Justice as just another cost of doing business. In this industry, anything and everything goes.
The logical question to ask is: “How does the FDA allows this to happen?” After all, the FDA is the regulatory agency assigned to oversee the drug industry.
Unfortunately, just like the doctors, the FDA is also under the influence of Big Pharma.
Here’s what you need to know.
1. A revolving door exists between Pharma and the FDA
Big Pharma has held sway over the FDA for many years, with FDA officials happy to “help” pharma, patient be damned. That’s because after their stint at the FDA, regulators know they can have a very lucrative career with the pharmaceutical companies, if they play the game properly.
Watch as attorney Mark Papantonio explains to Truth In Media’s investigative journalist Ben Swann about the revolving door between the FDA and the pharmaceutical companies.
The following is an example of what happens to patients when FDA regulators appease Big Pharma.
Zoloft, is one of several anti-depressants known to have some serious adverse events, including suicidal ideation. Drugwatch.com journalist Michele Lamas recounts the tragic effect the drug had on “Woody” Witczak, whose doctor, in 2003, prescribed Zoloft to help him sleep better. As per his wife, shortly thereafter he started experiencing adverse effects including
“diarrhea, night sweats, trembling hands, nightmares and worsening anxiety. Woody became highly agitated and irritable. His insomnia worsened. He described feeling like his head was outside his body.”
“He [their doctor] told us that we should give [Zoloft] four to six weeks to start working,” Kim said.
Five weeks later, Woody — a happily married, energetic, compassionate and cheerful man with a successful career — was dead. He had hung himself in the garage.
…
The family discovered Zoloft, an FDA-approved antidepressant, led to Woody’s tragic death.
No one warned Woody and his wife about the effects of the drug; as with everything else, they simply trusted their doctor. However, it was later revealed that:
Dr. Paul Leber, then director of FDA’s Division of Neuropharmacological Drug Products, knew there were problems with Zoloft’s efficacy data in 1991. But he “assured Pfizer that he thought he could convince the Committee that the studies were sufficient to recommend approval,” according to legal documents that surfaced in 2013.
One of the issues that Papantonio discusses in the video above is off-label prescribing, which a doctor is free to do, although a drug rep cannot legally recommend off-label use to a doctor. Why did his doctor prescribe an antidepressant for sleep issues, when Woody’s wife said that had no history of depression?
Learn how the FDA really evaluates psychiatric drugs here.
Learn about antidepressants causal relationship to suicides and homicides here, here, and here.
2. FDA advisory committee members are in Pharma's pocket
Drug approval is subject to the recommendations of an outside advisory panel to the FDA. Committee members include doctors and other scientific experts, consumer representatives, industry representatives, and FDA patient representatives. Investigative journalists Charles Piller and Jia You, writing for Science.org, reveal how advisors who vote to approve the drug, and whose pre-existing conflicts of interest the FDA ignores, are subsequently showered with financial opportunities and support. The following chart and illustration show how they are "compensated".
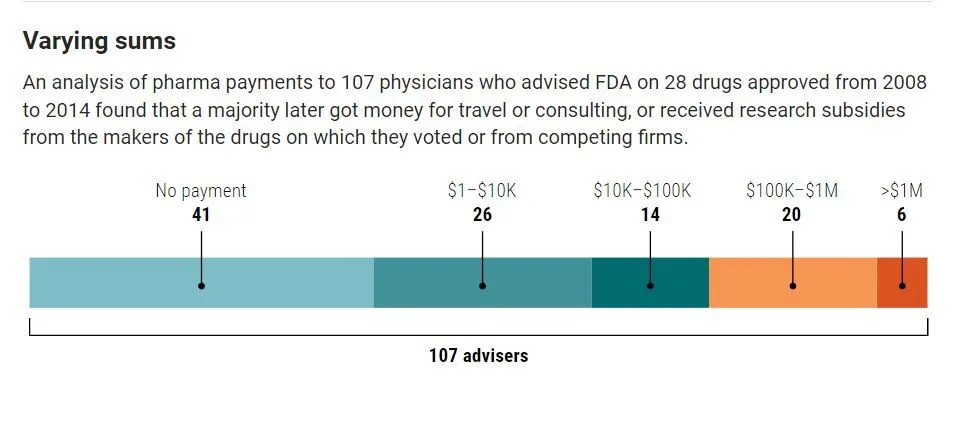
The illustration below shows how FDA advisors were rewarded/courted after approving a drug (which subsequently became a billion dollar blockbuster drug) that offers little improvement over existing medications of the same type while costing 25 times more.
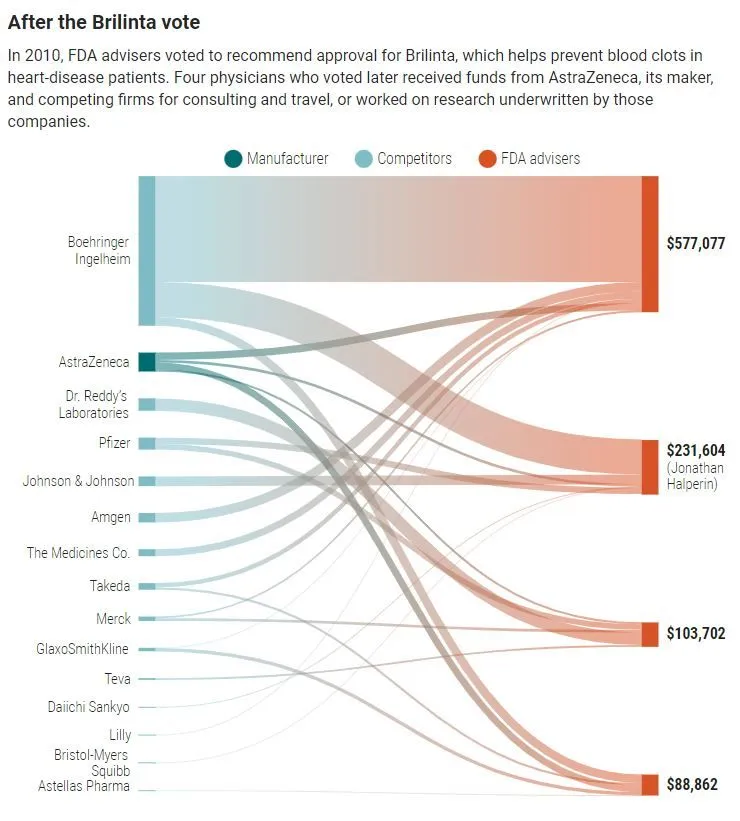
ProPublica writer Caroline Chen quotes a former FDA staffer who disturbingly articulated a consequence of such payments:
“You know who never shows up at the [advisory committee]?
The people who died in the trial,” … “Nobody is talking for them.”
3. The FDA is a captured agency
Regulatory capture is when an agency comes to identify with the industry it is charged to regulate. This is what happened to the FDA.
In 1992 Congress legislated the “unholy alliance” between the FDA and Big Pharma when it passed the Prescription Drug User Fee Act of 1992 (PDUFA) in response to complaints by drug companies and the public about the unduly long time that the agency was taking to review clinical trials and approve new drugs. The Act, as explained by Donald Light in his paper published in the Journal of Law, Medicine and Ethics, required the drug companies to pay user fees so the FDA could hire additional staff. In return, the FDA was supposed to speed up the approval process; priority applications would be reviewed within 6 months and regular applications within 12 months.
An in-depth analysis found that each 10-month reduction in review time — which could take up to 30 months — resulted in an 18.1-percent increase in serious adverse reactions, a 10.9-percent increase in hospitalizations, and a 7.2-percent increase in deaths.
Deadlines for drug approvals put agency staffers under significant stress; drugs approved within two months of the deadline had 3.27 times more black box warnings than drugs approved at other times and a 6.92 times greater likelihood that the drug would be withdrawn because of serious adverse events. Furthermore, concerns arising at the end of the review period were not adequately addressed. As a result, tens of thousands of additional people were hospitalized, had adverse reactions, and died.
Studies have shown, as recently reported by Marina Zhang for the Epoch Times, that of 1,567 drugs reviewed during the period 2008-2021, 94% lacked high-quality evidence of their benefits while harms were underreported. During the period 1983-2018 (even before the PDUFA was enacted) the FDA was approving more drugs with less rigorous testing, shortened review periods, and with substantial harms.
This is how it happens:
A. Pharma dictates to the FDA how to do its job
As part of the PDUFA, Congress required the FDA to negotiate the terms of the agreement with the pharmaceutical companies and since the PDUFA expires every five years, the FDA is forced to keep renegotiating the terms each time. Currently, the drug industry provides the FDA with 75-80% of it’s budget for scientific reviews. In return, the FDA does Pharma's bidding.
An investigative report by David S. Hilzenrath for the watchdog organization, POGO (The Project On Government Oversight), looks at the ways in which the PDUFA has enabled Big Pharma to dictate to the FDA, rather than the other way around. He wrote about PDUFA renewal negotiations for the period from September 2015 to January 2016, were closed to the public, and included teams of FDA officials in 70 meetings with drug company executives and lobbyists in order to set goals.
… [The negotiations] also entailed an extensive review and rethinking of how the FDA does its job. Tentatively, it has set the FDA on a course to increasingly look beyond data from controlled clinical trials—the costly mainstay of medical research—when assessing drugs. In the commitment letter, in buzzwords that track industry’s wish list, the FDA has pledged to pursue the use of “patient-reported outcomes” and “real-world evidence.” The agency also promised to explore “novel clinical trial designs,” including “highly innovative” ones for which “simulations are necessary.”
…
Unless “real-world evidence” follows principles of well-designed clinical trials, such as comparing the experimental drug to a placebo and committing to a study design before observing the results, “then it should be regarded as simply a deregulatory exercise and a departure from rigorous science,” Harvard’s [Daniel] Carpenter [a professor of government who has published work on the FDA and on “regulatory capture”] said by email. (Emphasis added.)
What did you believe about the way the FDA makes decisions regarding safety of the drugs you and your family and friends use?
B. FDA repays Big Pharma, Big Time, for its user fees
FDA repays pharma by approving dangerous drugs.
In her report, Caroline Chen, demonstrated how this happens with an account of FDA approval for two drugs, Nuplazid and Uloric, despite both being more dangerous and deadly than standard of care or no treatment. Both had failed two out of three clinical trials.
… Uloric’s manufacturer reported last November that patients on the drug were 34 percent more likely to die from heart disease than people taking an alternative gout medication. And since the FDA fast-tracked approval of Nuplazid and it went on the market in 2016 at a price of $24,000 a year, there have been 6,800 reports of adverse events for patients on the drug, including 887 deaths as of this past March 31.
The FDA is increasingly green-lighting expensive drugs despite dangerous or little-known side effects and inconclusive evidence that they curb or cure disease.
This has resulted in more and more unsafe and unproven drugs being approved for market, as she states:
The FDA okayed 46 “novel” drugs — whose chemical structure hadn’t been previously approved — in 2017, the most in at least 15 years. At the same time, it’s rejecting fewer medications… (Emphasis added.)
Furthermore,
The FDA also increasingly allows drugmakers to claim success in trials based on proxy measurements — such as shrunken tumors — instead of clinical outcomes like survival rates or cures, which take more time to evaluate. In return for accelerated approval, drug companies commit to researching how well their drugs work after going on the market.
Are drug companies saving money on drug trials by making the public unwitting and unconsenting trial participants?
Are drug companies earning profits from negligently approved products while destroying the public's health?
Proxy measurements, also called surrogate endpoints (the FDA has a bunch of approved ones here), should never be used a substitute for clinical outcomes. In a PBS report about pharma and the FDA, Raymond Woosley, who had been a top candidate for FDA commissioner in 2002, explained how using surrogate endpoints can have deadly consequences.
Even a chair of an FDA drug review committee is disturbed by the FDA's constant approval of dangerous drugs. For over 25 years the FDA has continuously approved ever more dangerous opioid drugs (see the bottom of section 3). In an article highlighting the issue Matt Agorist, Editor at Large at the The Free Thought Project, related the impact that the FDA's ignoring the opioids' harms has had on Dr. Raeford Brown, chair of the FDA’s committee that reviews opioid based drugs prior to approval. Brown said that he had lost faith in the agency's ability to protect the public. The FDA, Brown said, is more interested in kowtowing to Big Pharma and described the FDA's loyalty to the drug industry as potentially criminal.
Listen to Russel Brand read about serious malfeasance at the FDA.
Read more here, here, here, and here.
C. The PDUFA enabled industry to subvert the FDA
Following are some examples.
- A 2006 assessment of the FDA by the Institute of Medicine, at the FDA’s request, included the following:
[The PDUFA] has had some drawbacks, including increasing the agency’s dependence on industry funding for its drug review activities, severely skewing CDER’s focus to facilitating review and approval perhaps at the expense of other center activities, and creating an environment of intense pressure on its reviewers (Zelenay, 2005)
- From Hilzenrath's investigative report:
- The culture of the FDA “views the pharmaceutical industry it is supposed to regulate as its client. It overvalues the benefits of the drugs it approves, and it seriously undervalues, disregards and disrespects drug safety,” [FDA safety official David] Graham said at the 2004 Senate hearing.
- [Dr. Ellis] Unger, director of an FDA drug evaluation office, said the case for approval [of Sarepta, a drug being trialed for Duchene Muscular Dystrophy] was weak.
“The approval of this NDA”—new drug application—“in its present form would have far reaching negative consequences for the public health,” he wrote in a July memo.Unger was overruled by [Janet} Woodcock [head of the FDA’s Center for Drug Evaluation and Research]. He appealed to an internal FDA board responsible for resolving scientific disputes.In a presentation to the board, Woodcock talked about the drug maker’s financial condition and stock price. “Dr. Woodcock cautioned that, if Sarepta did not receive accelerated approval for eteplirsen, it would have insufficient funding to continue to study eteplirsen and the other similar drugs in its pipeline,” Luciana Borio, the FDA’s acting chief scientist, wrote in an account of the appeal.Woodcock also “emphasized her view that the agency needs to accept more uncertainty when granting accelerated approval,” Borio wrote. (Emphasis added.)
- From Chen's report:
The more that the FDA relied on industry fees to pay for drug reviews, the more it showed an inclination towards approval, former employees say.
“You don’t survive as a senior official at the FDA unless you’re pro-industry,” said Dr. Thomas Marciniak. A former FDA medical team leader, and a longtime outspoken critic of how drug companies handle clinical trials, Marciniak retired in 2014. “The FDA has to pay attention to what Congress tells them to do, and the industry will lobby to get somebody else in there if they don’t like you.”
Staffers know “you don’t get promoted unless you’re pro-industry,” he added.
- A letter written to the FDA in October 2021, by Michael T. Abrams for the Public Citizen’s Health Research Group, regarding the upcoming renewal of the PDUFA included the following:
- … Aaron Kesselheim M.D., J.D., M.P.H., Director of the Division of Pharmacoepidemiology and Pharmacoeconomics at Harvard Medical School, resigned from the Peripheral and Central Nervous System Advisory Committee because of the FDA’s decision to approve aducanumab (Aduhelm) for treatment of Alzheimer’s disease in direct opposition to the nearly unanimous opinion of that expert advisory committee. In his resignation letter to Acting FDA Commissioner Janet Woodcock, Dr. Kesselheim wrote: “FDA is not presently capable of adequately integrating the Committee’s scientific recommendations into its approval decisions.” (pp 1-2)
- … research that shows that 81% of NDA [new drug approvals] approvals in 2018 involved Accelerated Approval, Fast-Track, or Priority Review8 and that faster approvals under PDUFA correlated with the marketing of products that were less safe than those marketed before the PDUFA was instituted.9
- … interactive communications established under PDUFA have resulted in inappropriately close collaborations between the Agency and sponsors that have compromised the integrity for NDA/BLA [biologics license applications] reviews. (p4) (Emphasis added.)
4. Pharma embedded in FDA-affiliated nonprofit
A little-known foundation, the Reagan-Udall Foundation for the FDA (RUF), is a non-profit corporation created by Congress in 2007 to "… advance the mission of the Food and Drug Administration to modernize medical, veterinary, food, food ingredient, and cosmetic product development, accelerate innovation, and enhance product safety.” (Both the NIH and CDC have their own non-profits funded by Big Pharma.)
Just as Congress wrote the PDUFA to favor the pharmaceutical industry, so too did it set up the RUF to favor the industry. Ex officio members were to be members of agencies already beholden to Big Pharma: the [FDA] Commissioner, the Director of the NIH, the Director of the CDC, and the Director of AHRQ (the Agency for Healthcare Research and Quality).
Those members would appoint 14 additional board members: 9 from a list of candidates to be provided by the National Academy of Sciences and 5 from lists of candidates provided by patient and consumer advocacy groups, professional scientific and medical societies, and industry trade organizations. They are to include 4 representatives from the general pharmaceutical, device, food, cosmetic, and biotechnology industries, 3 from academic research organizations, 2 from patient or consumer advocacy organizations, 1 from health care providers, and 4 at-large members with expertise or experience relevant to the purpose of the Foundation.
The Foundation’s Policies webpage states that
“… our bylaws outline the conflict of interest policies that have been put into place to ensure that no one entity or organization has undue influence on Reagan-Udall Foundation initiatives and projects.
The Reagan-Udall Foundation has come under scrutiny for conflicts of interest.
A 2012, NPR article by Nell GreenfieldBoyce reflects the concerns of those who are wary of the Foundation’s funding sources such as the FDA as well as industry related organizations, such as the PhRMA Foundation, a nonprofit funded by pharmaceutical companies. PhRMA provided Reagan-Udall with a $150,000 grant that helped it get off the ground; funding from the FDA had been held up due to the fears of conflict of interest that such funding presented.
According to the article, the Foundation said, earlier on, that they would accept grants for its core operating expenses from the government, individual donors, or other nonprofits, but not from industry. Interestingly enough (because of the requirement that members of industry sit on the board), Garry Neil, who is an executive at Johnson & Johnson and chairman of the PhRMA Foundation's board of director, and is also a member of Reagan-Udall's board, claimed that it was fine for Reagan-Udall to take direct support from the PhRMA Foundation, which he pointed out was another nonprofit.
"It's a foundation, so the money originally came from industry. But it's now in the foundation, and it's a nonprofit philanthropic organization that funds high-quality, peer-reviewed science," Neil says.
However, GreenfieldBoyce quotes Sidney Wolfe, a consumer advocate at Public Citizen, who pointed out that
… the PhRMA Foundation might technically be a nonprofit, "but one can hardly expect that they're going to do things that are not in the interests of their funders."
"The FDA is already overly and dangerously influenced by the drug industry, the medical device industry, the food industry," says Wolfe, who points out the agency gets huge amounts of money in user fees from the companies that make the products the agency reviews.
GreenfieldBoyce also noted that the Reagan-Udall Foundation received $1 billion from the Bill and Melinda Gates Foundation (BMGF) for projects related to tuberculosis.
She quotes Francesca Grifo of the Union of Concerned Scientists as stating that
"[t]here's been an enormous amount of research that shows us that funding influences decisions, and that's a problem," Grifo says.
It is notable that the RUF Bylaws, above, which stated that they would ensure that no one entity or organization would have undue influence over the RUF, never addressed the undue collective influence a conglomeration of entities representing a single industry would have.
It is important to note that the BMGF is one of the largest funders of the WHO. As a non-state donor, it can earmark its donations for pet projects; counting WHO funding by BMGF funded/directed organizations, Gates is effectively subverting the WHO’s mission.
In 2020, Reagan-Udall received a $250,000 grant from the Rockefeller Foundation for a COVID-19 Diagnostics Evidence Accelerator. Learn how Rockefeller funding was responsible for shaping mainstream medicine here and here.
Others are also concerned about the cozy relationship between the Foundation and pharma. CrossFit, a program focused on assisting people to improve their health, is enraged by industry influence,
In August 2019, CrossFit railed against the agencies entrusted with America’s health for having failed to provide meaningful answers and solutions to the rising rates of chronic disease and illness, decried the influence that the pharmaceutical and food/beverage industries have on them, and on the Reagan-Udall Foundation in particular.
Public-private partnerships, facilitated by a government agency’s affiliated foundation, can compromise the integrity of that agency’s mission, especially in the absence of transparency. The only way to ensure the veracity of the science and research coming from an organization, especially one with such attractive proximity to government influence and authority, is to know who is providing the funding and whether there are any restrictions on its expenditure.
On May 2, 2019, CrossFit, Inc. lobbyist Brett Ewer spoke at the annual meeting of the FDA’s Reagan-Udall Foundation (RUF) and demanded that RUF leadership fully produce annual funding reports, which by law must provide a specific accounting of the sources and uses of its funds. As of the date of this publication, the RUF has not produced a report meeting those requirements. (Emphasis added.)
The image below is of the donor sponsorships from the RUF 2021 annual report. It is easy to see that they are heavily weighted towards the pharmaceutical industry. Major donors include Garry Neil and PhRMA.

FDA and the Reagan-Udall Foundation work hand-in-hand
The opioid crisis is a case-in-point of how the FDA and the Reagan-Udall Foundation work together to support pharma. About 50,000 people die from opioid overdoses in the US annually.
- Three regulatory failures on the part of the FDA, as explained by Dr. Andrew Kolodny, led to the opioid crisis. Dr. Kolodny is a senior scientist and medical director of Brandeis' Opioid Policy Research Collaborative and is recognized as one of the country's top addiction specialists.
- FDA Principal Deputy Commissioner Janet Woodcock was complicit in fueling the opioid crisis during her 23 year history as chief of the FDA's CEDR (Center for Drug Evaluation and Research), what Kolodny called the “the worst medical regulatory failure in U.S. history.” Gerald Posner, opinion contributor to USA Today, reviewed her role in this crisis.
- The FDA attacked Kratom, an herbal plant in the coffee family that has been found to be a safe and effective by millions of people looking to get off opioids and/or for safer and milder pain relief. As Agorist wrote for The Free Thought Project, the FDA lied by calling it an opioid and a dangerous drug. Kratom, unlike opioids (synthetic opiates), does not depress the respiratory system, the reason that people overdosing on opioids die.
FDA Commissioner Scott Gottlieb “claimed that he and the FDA are acting “in the interest of protecting public health.” "Gottlieb was a member of GlaxoSmithKline’s product investment board. … GlaxoSmithKline owns a patent on a kratom alkaloid designed for the very purpose of treating pain, thereby alleviating dependency on opioids.”
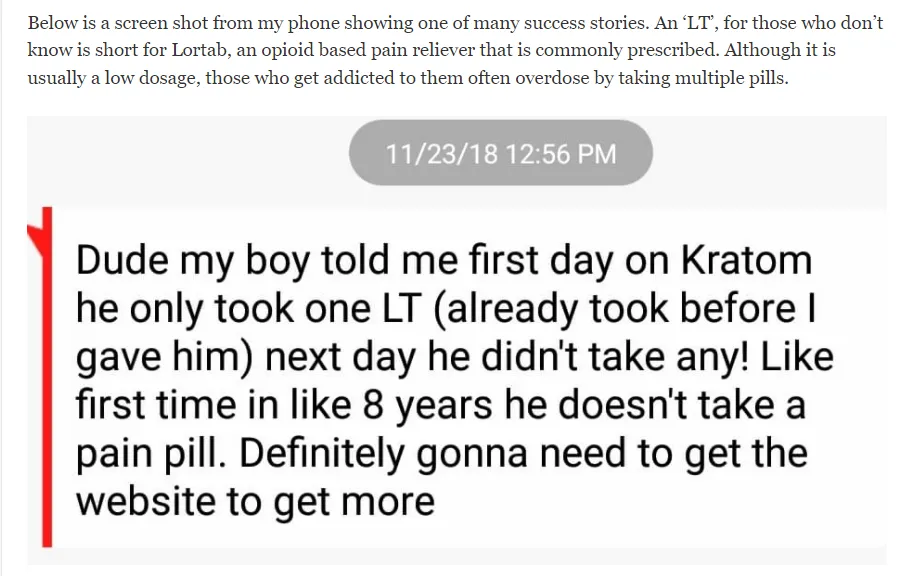
People are grateful to transition from opioids to Kratom as is evidenced by the message in the image below.
- The Reagan-Udall Foundation held a virtual public meeting to “help answer some of the most frequently asked questions about access to naloxone and hear from individuals, practitioners, advocates, and others about their experiences with naloxone access.”
Naloxone, sold under the brand name Narcan, among others, is a drug that reverses depression of the respiratory system in opioid drug overdoses. Naloxone, according to drugs.com:
“… may precipitate abrupt opioid withdrawal in physically dependent persons; signs and symptoms may include body aches, fever, sweating, sneezing, yawning, nausea, vomiting, sweating, lacrimation, rhinorrhea, cramping, insomnia, chills/hot flashes, piloerection, tachycardia, anxiety, restlessness, irritability, tremulousness, hypertension, seizures, and cardiac arrest. In the neonate, opioid withdrawal may also include convulsions, excessive crying, and hyperactive reflexes.” (Emphasis added.)
Under respiratory effects are dyspnea - difficulty breathing resulting in shortness of breath, respiratory depression - the very problem the drug is supposed to correct, and hypoxia - lack of oxygen to parts or a part of the body, such as the brain.
According to SAMSHA (Substance Abuse and Mental Health Services Administration) medical attention must be sought as soon as possible after administering/receiving the drug.
In December 2021, the WHO’s Executive Committee on Drug Dependency (ECDD) "… concluded that there is insufficient evidence to recommend a critical review of kratom."
Kyle Jaeger, reported on the ECDD decision for Marijuana Moment:
“People report using kratom to self-medicate a variety of disorders and conditions, including pain, opioid withdrawal, opioid use disorder, anxiety and depression,” ECDD said in its report. “Kratom is being used as a part of traditional medicine in some countries.”
Included in the report is this message from Mac Haddow, senior fellow on public policy for the American Kratom Association (AKA) discussing the decision:
A Google search shows that naloxone is widely promoted as a life-saving drug, prescribed for emergency use to patients and families of patients who are at high risk of drug overdose.
It seems obvious that allowing Kratom is a better choice than waiting for people to overdose on opioids so they need naloxone.
Read more here, here, and here.
5. FDA legitimizes drugs for pharma-fabricated diseases
Donald W. Light, in his article published by Harvard's Edmond J. Safra Center for Ethics, demonstrates how the FDA, by lowering and widening criteria, has enabled industry to make up new diseases, essentially medicalizing normal.
He provides a “pre-Alzheimer's” drug as an example.
… The New England Journal of Medicine has published, without comment, proposals by two senior figures from the FDA to loosen criteria [for] drugs that allege to prevent Alzheimer’s disease by treating it at an early stage.8 … the FDA incorporate[s] the language and rationale of marketing executives for the industry. First, they use the word “disease” to refer to a hypothetical “early-stage Alzheimer’s disease” that supposedly exists “before the earliest symptoms of Alzheimer’s disease are apparent.” Notice that phrasing assumes that the earliest symptoms will become apparent, when in fact it’s only a hypothetical model for claiming that cognitive lapses like not remembering where you put something or what you were going to say are signs of incipient Altzheimer’s [sic] disease. The proposed looser criteria would legitimate drugs as “safe and effective” that have little or no evidence of being effective and expose millions to risks of harmful side effects.
No proven biomarkers or clinical symptoms exist, the FDA officials note, but nevertheless they advocate accelerated approval to allow “drugs that address an unmet medical need.” What “unmet need"? None exists. This market-making language by officials who are charged with protecting the public from unsafe drugs moves us towards the 19-century hucksterism of peddling cures of questionable benefits and hidden risks of harm, only now fully certified by the modern FDA.9
The main reason for advocating approvals of drugs for an unproven need with unproven benefits, these FDA officials explain, is that companies cannot find effective drugs for overt Alzheimer’s. (Emphasis added.)
… The job of the FDA, it seems, is to help drug companies open up new markets to increase profits for the FDA’s corporate paymasters.
… Similar FDA-encouraged shifts have been made for drugs treating pre-diabetes, pre-psychosis, and pre-bone density loss, with few or no benefits to offset risks of harm.
Have you been prescribed medications for a disease that you don't have?
6. FDA ignores false and fabricated trial data it discovers
New York University journalism professor Charles Seife and students in his investigative reporting class searched through FDA documents to uncover what the FDA knew about fraudulent drug trials but wasn't telling anyone. He recounted their findings (documented in a report published in JAMA) in an article for Slate Magazine. What they discovered presents a scathing criticism of the FDA’s watchdog practices.
Among other things, they discovered that FDA inspections of clinical trials have revealed tremendous amounts of fraud, including “Faked X-ray reports. Forged retinal scans. Phony lab tests. Secretly amputated limbs” yet none of these violations are reported.
Seife explained how far they went to hide the evidence:
… For more than a decade, the FDA has shown a pattern of burying the details of misconduct. As a result, nobody ever finds out which data is bogus, which experiments are tainted, and which drugs might be on the market under false pretenses. The FDA has repeatedly hidden evidence of scientific fraud not just from the public, but also from its most trusted scientific advisers, even as they were deciding whether or not a new drug should be allowed on the market… For an agency devoted to protecting the public from bogus medical science, the FDA seems to be spending an awful lot of effort protecting the perpetrators of bogus science from the public. (Emphasis added.)
The agency was also meticulous in making sure that no one looking at the documents would be able to figure out the truth, either.
When there are problems, the FDA generates a lot of paperwork—what are called form 483s, Establishment Inspection Reports, and in the worst cases, what are known as Warning Letters. If you manage to get your hands on these documents, you’ll see that, most of the time, key portions are redacted: information that describes what drug the researcher was studying, the name of the study, and precisely how the misconduct affected the quality of the data are all blacked out. These redactions make it all but impossible to figure out which study is tainted.
Here is an image from one document he gave as an example (retrieved from the Wayback Machine here):

Of the roughly 600 clinical trials with a failed FDA inspection, for only about 100 of them were Seife and his students able to figure out which study, drug, and pharmaceutical company was involved. They uncovered what was redacted by cross-referencing the documents with data from clinical trials, checking a variety of other databases, and using carefully crafted Google searches.
The FDA overlooks falsified data in generic drugs, also
It’s not just fraud related to brand name drugs that the FDA looks away from. The agency also ignores falsification of data by the manufacturers of generic drugs.
This was exposed by investigative journalist Katherine Eban, in her book Bottle of Lies: The Inside Story of the Generic Drug Boom, published in 2019. This is important because 90% of all prescriptions filled in the U.S. are for generic drugs, predominantly manufactured in India and China.
She discusses what she uncovered in the following interview. (This is part 2 of the interview; part 1 can be found here.)
A more extensive interview can be seen here and her TedTalk here.
Jim Edwards, in his 2009, blog post for the online business magazine Bnet, revealed that the FDA continued approving drugs by Indian manufacturer Ranbaxy even though it found out that the firm had faked test results it sent to them!
Where is the public and Congressional outrage? How has this been allowed to continue? Do your state representatives know about this?
The public must be proactive
Here's what you can do:
Learn about the medications you you are currently taking and about those your doctor wants to prescribe to you.
Doctors recommend waiting between 5-10 years before using a new drug, unless no other of its kind is available.
Let your representatives and the FDA know that putting patients in danger by approving unsafe and ineffective drugs is not acceptable. There must be accountability.
Forward this article to your friends and family so that they can be informed, too.


.webp)

